Biosimilar Approval: How FDA Reviews Biologic Alternatives in 2025
 Dec, 1 2025
Dec, 1 2025
The U.S. Food and Drug Administration (FDA) doesn’t approve biosimilars the same way it approves generic pills. That’s because biosimilars aren’t simple chemical copies-they’re complex, living-molecule drugs made from living cells. Think of them as close relatives, not twins, of expensive biologic drugs used to treat cancer, rheumatoid arthritis, diabetes, and Crohn’s disease. While a generic version of aspirin is chemically identical to the brand-name version, a biosimilar must be shown to be highly similar-with no meaningful differences in safety or effectiveness-despite tiny, unavoidable variations that come from using biological systems to make them.
Why Biosimilars Are Different from Generics
Generics are made from synthetic chemicals. Their structure is simple, predictable, and easy to replicate. Biosimilars, on the other hand, are produced in living cells-usually yeast or hamster ovary cells-using intricate processes. Even small changes in temperature, pH, or cell culture conditions can alter the final product. That’s why the FDA requires hundreds of analytical tests to compare a biosimilar to its reference biologic. These tests look at protein structure, sugar attachments, purity, and how the molecule behaves in the body. No single test is enough. It’s the full picture that matters.The old approval process required a full clinical trial comparing the biosimilar to the original biologic in patients to prove it worked the same way. That meant years of testing, millions of dollars, and delays that kept patients from accessing cheaper options. In October 2025, the FDA changed that.
The 2025 FDA Guidance: A Major Shift
The FDA’s new draft guidance, released on October 29, 2025, is the biggest update to biosimilar rules since the Biologics Price Competition and Innovation Act (BPCIA) passed in 2010. It removes the automatic requirement for comparative clinical efficacy studies. Now, if a biosimilar passes deep analytical testing, shows matching pharmacokinetics (how the body absorbs and processes the drug), and has no red flags in immunogenicity (the risk of triggering an immune response), the FDA may approve it without running a full patient trial.This shift is based on science, not politics. Modern tools like mass spectrometry and advanced chromatography can now detect differences at the molecular level that were invisible just a decade ago. The FDA says these tools are so precise, they can predict clinical outcomes better than small, short-term patient studies. For well-understood drugs like adalimumab (Humira) or trastuzumab (Herceptin), where the link between molecular structure and patient response is clear, this approach is a game-changer.
Companies that used to spend $200 million and eight years to get a biosimilar approved might now do it in five years for under $150 million. That’s a big deal when the reference biologic costs $50,000 to $100,000 per patient per year.
Interchangeability: What It Means and Why It’s Controversial
Interchangeability is the holy grail for biosimilars. It means a pharmacist can swap the biosimilar for the brand-name drug without asking the doctor. In 2025, the FDA approved two denosumab biosimilars-Enoby and Xtrenbo-with interchangeability status. That was the first time multiple interchangeable biosimilars were approved for the same reference product.But here’s the twist: FDA Commissioner Marty Makary said at a conference in October 2025, “Every biosimilar should have the designation of interchangeable.” That’s not what the law says. Under current U.S. law, interchangeability requires extra data-switching studies-that prove patients can alternate between the reference and biosimilar without increased risk. The FDA has been granting this status on a case-by-case basis, but Makary’s comments suggest the agency is pushing for a broader interpretation.
Some experts agree. Dr. Mark Eisner, former FDA deputy director, called the new guidance “the most scientifically sound approach yet.” Others worry. Dr. Paul Baldrick of the Journal of Biological Sciences warned that subtle differences might only show up after years of use in chronic conditions. And Dr. Robert Popovian from PhRMA pointed out that removing the interchangeability distinction without Congress changing the law could confuse doctors and pharmacists.
Right now, 34 states still require prescriber approval before a pharmacist can switch a patient to a biosimilar. That’s a major barrier. Even if the FDA says a drug is interchangeable, state laws can block it.

Who’s Making Biosimilars and Who’s Using Them
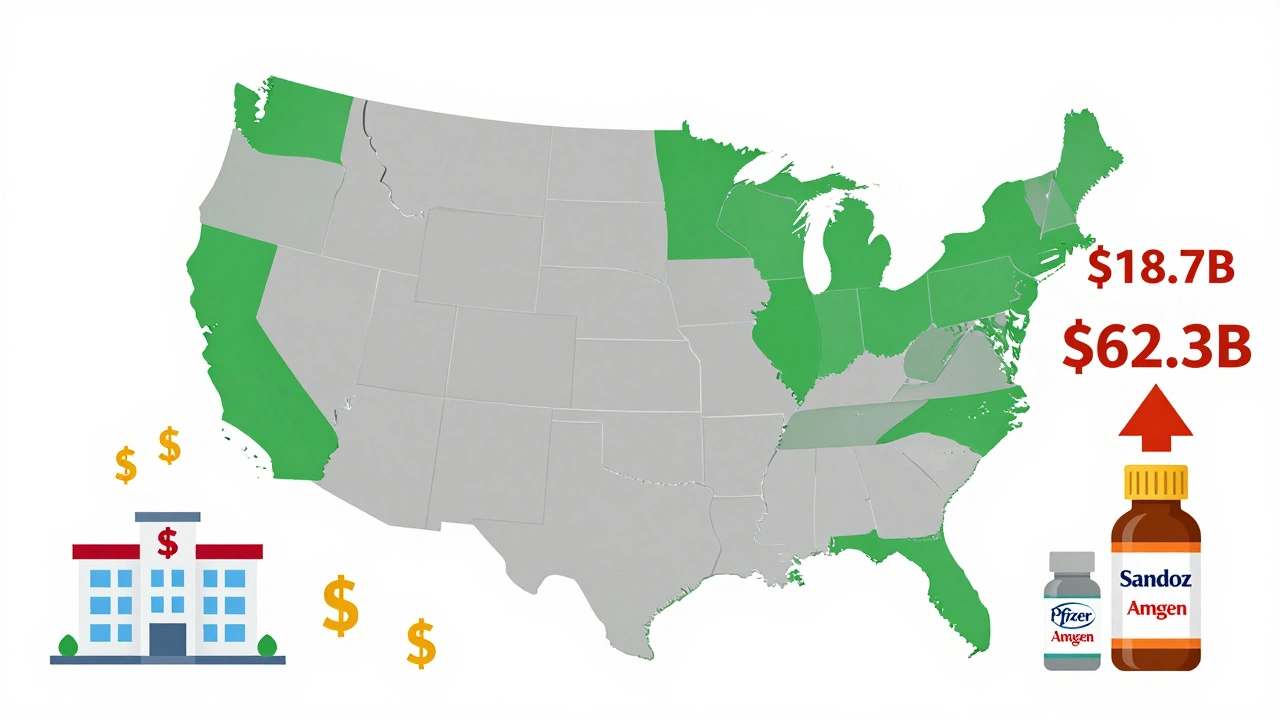
As of late 2025, the FDA has approved 76 biosimilars. But only 28 companies have brought one to market. Big players like Sandoz, Pfizer, and Amgen dominate, with 17, 12, and 10 approvals respectively. Smaller biotechs struggle. The analytical tools and testing infrastructure needed are expensive. Only 12 of the 76 approved biosimilars came from companies with fewer than 100 employees.But things are changing. Viatris and Biocon are gaining ground. And the market is growing fast. The U.S. biosimilar market was worth $18.7 billion in 2024 and is projected to hit $62.3 billion by 2029. Hospitals are already seeing results. Mayo Clinic reported a 37% drop in biologic drug costs after switching to biosimilars for cancer treatments-saving $18 million a year.
But patient awareness is low. Only 32% of patients know what a biosimilar is, according to the National Biosimilars Survey. On Reddit’s r/pharmacy, patients switching to biosimilars for rheumatoid arthritis reported mostly positive experiences-63% said efficacy was the same. But 22% noticed minor side effects, like more injection site redness. Concerns about safety were common at first, but 68% of those worries disappeared after talking to their doctor.
The Road Ahead: Challenges and Opportunities
The FDA’s new guidance is a step forward, but hurdles remain. Patent lawsuits delay biosimilar entry in 68% of cases, according to the FTC. Companies spend years in court fighting to keep prices high. Even when a biosimilar is approved, it can’t reach patients if the original drug maker holds onto patents.Another issue is complexity. Biosimilars for simple monoclonal antibodies are easier to develop. But for antibody-drug conjugates-drugs that attach a toxin to an antibody to kill cancer cells-the link between structure and effect is less clear. The FDA’s guidance acknowledges this, saying more data may still be needed for these complex products.
The FDA’s Biosimilars User Fee Amendments of 2022 (BsUFA III) are helping. The program funds faster reviews and gives companies clearer timelines. The first BPD meeting fee is due within seven days after the FDA agrees to meet. Annual fees continue through September 2027.
Support is growing. The FDA’s Biosimilars Community Resource Center had over 12,700 visitors in October 2025. The Biosimilars Council has provided 87 free consultations to small developers since 2023.
Europe has been ahead of the U.S. for years. The EMA has approved over 100 biosimilars since 2006. In Europe, biosimilars make up 67% of the market for comparable biologics. In the U.S., it’s only 23%. The 2025 guidance aims to close that gap. Analysts predict U.S. market share could jump to 40-50% by 2030, saving the system up to $150 billion a year.
The real test won’t be how many biosimilars get approved. It’ll be whether patients, doctors, and pharmacists trust them enough to use them.
What This Means for Patients
If you’re on a biologic drug that costs thousands of dollars a month, a biosimilar could cut that cost by 50% or more. But you won’t automatically get one. You have to ask. Talk to your doctor. Ask if a biosimilar is right for you. Check with your pharmacy. Ask if your state allows automatic substitution.Don’t assume a biosimilar is “less effective.” The science says it’s not. The FDA doesn’t approve anything that isn’t as safe and effective as the original. The difference is cost-and access.
For chronic conditions like rheumatoid arthritis or Crohn’s disease, where patients take the drug for years, even small savings add up. For cancer patients, it can mean the difference between staying on treatment or dropping out because of cost.
The FDA’s 2025 update is a turning point. It’s not perfect. But it’s a move toward making life-saving drugs affordable. The next step? Congress needs to fix the interchangeability rules. And patients need to speak up.
Are biosimilars as safe as the original biologic drugs?
Yes. The FDA requires biosimilars to meet the same high standards for safety, purity, and potency as the original biologic. Approval is based on hundreds of analytical tests, pharmacokinetic data, and immunogenicity studies. No meaningful differences in safety have been found in approved biosimilars. Post-market monitoring continues to track any rare side effects.
Can a pharmacist switch my biologic to a biosimilar without my doctor’s permission?
It depends on your state. The FDA can designate a biosimilar as interchangeable, meaning pharmacists can substitute it without a new prescription. But 34 states still require the prescriber to approve the switch. Even if the FDA says it’s interchangeable, your state’s law controls what pharmacists can do. Always check with your pharmacy and doctor.
Why are biosimilars cheaper than biologics?
Biosimilars cost less because they don’t need to repeat the original clinical trials that proved the biologic works. The 2025 FDA guidance allows companies to rely on advanced analytical data instead of lengthy patient studies. That cuts development time from 8-10 years to 5-7 years and reduces costs from $200 million to under $150 million per product. These savings are passed on to patients and insurers.
How many biosimilars has the FDA approved as of 2025?
As of late 2025, the FDA has approved 76 biosimilars for use in the U.S. These treat conditions including cancer, autoimmune diseases like rheumatoid arthritis, diabetes, osteoporosis, and inflammatory bowel disease. The number is expected to rise to 15-20 approvals per year under the new 2025 guidance.
Is the FDA’s new guidance final?
No. The October 2025 guidance is a draft and is open for public comment until January 27, 2026. The final version is expected by June 2026. While the draft represents the FDA’s current thinking, the final rules could include minor changes based on feedback from industry, patients, and healthcare providers.
What Comes Next?
The next five years will be critical. If patent barriers fall and interchangeability rules get clarified, biosimilars could transform how we pay for biologic drugs. Hospitals, insurers, and patients will benefit. But it won’t happen unless doctors feel confident prescribing them and patients feel safe using them.The science is ready. The FDA is ready. Now it’s up to the system to catch up.
Courtney Co
December 1, 2025 AT 23:04Priyam Tomar
December 3, 2025 AT 15:28Irving Steinberg
December 4, 2025 AT 06:31Lydia Zhang
December 4, 2025 AT 23:37Kay Lam
December 5, 2025 AT 09:03Eric Vlach
December 7, 2025 AT 07:52Souvik Datta
December 7, 2025 AT 14:42Matt Dean
December 7, 2025 AT 23:42Bee Floyd
December 9, 2025 AT 05:56Jeremy Butler
December 9, 2025 AT 09:49Shashank Vira
December 10, 2025 AT 17:18